研究报告/Research Report
萝卜叶绿体及线粒体基因组测序与组装 
 作者
作者  通讯作者
通讯作者
《分子植物育种》网络版, 2016 年, 第 14 卷, 第 2 篇 doi: 10.5376/mpb.cn.2016.14.0002
收稿日期: 2016年01月13日 接受日期: 2016年02月20日 发表日期: 2016年02月25日
引用格式(中文):
段乃彬等, 2016, 萝卜叶绿体及线粒体基因组测序与组装, 分子植物育种(online), 14(2): 1008-1015 (doi: 10.5376/mpb.cn.2016.14.0002)
引用格式(英文):
Duan et al., 2016, Assembling and Sequencing of Chloroplast and Mitochondrial Genomes of Radish (Raphanus Sativus L), Fenzi Zhiwu Yuzhong (online) (Molecular Plant Breeding), 14(2): 1008-1015 (doi: 10.5376/mpb.cn.2016.14.0002)
为从分子水平上探讨萝卜雄性不育机理及育性恢复关系,本研究对4个萝卜雄性不育系及1个萝卜雄性不育保持系材料进行了线粒体及叶绿体的基因组测序、组装。测序采用PE90策略,获得Clean data有1.2 G bp。通过数据过滤、计算测序深度及Kmer频度分析,确定了组装的mer值及覆盖度参数。我们先用Clean data组装叶绿体基因组,再用过滤的数据组装线粒体基因组,最后利用Sanger测序填补Gap来得到各基因组全长。结果表明参试样品萝卜叶绿体基因组长度在153 352~153 445 bp之间,线粒体基因组长度在239 696~258 853 bp之间,均包括1个LSC,1个SSC及1对反向重复序列。基因注释结果表明叶绿体基因组编码了87个蛋白基因、20个tRNA基因及4个rRNA基因,线粒体基因组则编码了82个蛋白基因,17个tRNA基因及3个rRNA基因。
研究背景
萝卜(Raphanus sativus L.)是重要的十字花科蔬菜(http://frps.eflora.cn/frps/Raphanus%20sativus),在东亚及南亚地区有着较大的栽培面积。作为异花授粉作物,萝卜具有显著的杂种优势。育种者为提高杂交制种纯度,在萝卜上利用雄性不育系进行育种。Ogura于1968年首先发现了胞质雄性不育类型(Ogura, 1968),我国育种者也从不同的地方品种中发现了萝卜雄性不育株,均属于Ogura不育类型(陈黎明等, 2009; 程计华等, 2008)。然而,Ogura胞质雄性不育机理目前尚不明确。此外,我们发现,同属于Ogura不育类型的品种,其保持和恢复关系仍有一定差别。研究表明胞质雄性不育与叶绿体或线粒体基因组的突变或结构变异密切相关。
另一方面,进行叶绿体、线粒体基因组组装,在研究植物进化、分类、遗传多样性等方面有重要的意义。而对基因组进行基因注释,是探讨植物光合作用机理、植物能量代谢、植物抗氧化及次生代谢的基础。
为揭示萝卜雄性不育的不育机理,并为植物细胞器基因组研究提供基础数据。我们选择了3个来源不同、并且保持关系有一定差别的Ogura不育材料、1个未知类型不育材料及1个具有正常细胞质的材料,进行了线粒体和叶绿体基因组的测序、数据过滤、Kmer分析、组装及基因注释。以期通过比较不同类型材料基因组间的序列差异,寻找与育性有关的基因及SNP变异。
1结果与分析
1.1测序数据Raw data和Clean data分析
本研究的5个萝卜样品测序的下机数据均为Fastqc格式,数据读长为90 bp,共获得Raw data有18 108 266对Reads。根据对Raw data的质控检验,GC含量均为42.5%。经去除测序重复、Adaptor及测序污染后,最终得到Clean data为1.25 G (表1)。
|
Table 1 Count of Raw data and clean data obtained in this research |
.png)
1.2数据过滤及各组分的比率
以NCBI公布的萝卜基因组组装序列(NCBI Accession Number: GCA_000715565.1)及萝卜的叶绿体、线粒体基因组为参考基因组(NCBI Accession Number: KJ716483, AB694744.1),进行BWA比对,通过对Sam文件进行数据过滤,分别得到了叶绿体、线粒体的测序数据(表2)。
|
Table 2 The composition and ratios of clean data |
.png)
5个供试样品的叶绿体测序深度均较高,平均值为858.3,而线粒体测序深度平均为117.2。分析结果表明,在叶绿体和线粒体的DNA提取过程中,已经去除了绝大多数细胞核基因组组分,核基因组的平均覆盖度仅为0.2。
1.3基于Kmer频度分析过滤样品测序数据
利用Jellyfish软件对5个样品的原始数据进行21mer分析,未见高频度的Kmer分布。估测的分子量大小为8 234 k,是预期线粒体,叶绿体基因组的40倍和55倍。推测此时有部分核基因组片段可以形成弥散分布的Kmer,而导致无法正确判断分子量。这样的数据若直接用于基因组的组装拼接,会形成冗余的Contigs。由于这些Contigs来自三个组分,并不是1个整体,不能组装到1个完整的基因组(图1)。
|
Figure 1 Kmer frequency distribution of RS41’s raw data |
.png)
经软件过滤后,对Reads进行21mer的Kmer分析可见明显的高频度组分,以41样品为例:叶绿体Reads,Kmer分析表明呈现两个明显的尖锐的峰(图2),即该数据存在两个组分,针对第1个峰计算的测序深度为,第二个峰计算的测序深度为,接近第1个组分的两倍,推测是1个重复序列。从数据量上看,第一组分中占有的比例是6/8,大约占有碱基量92 637 405,该数值除测序深度820可得该组分分子量为110 k。第二组份占有的比例是2/8,大约占有碱基数是30 879 135,该数值除测序深度1 600,得该组分子量约为20 k,合计分子量为该数值符合预期的萝卜叶绿体基因组碱基数。针对线粒体组装的Reads,其Kmer分布同样呈现明显的双峰(图3),比例关系为(26:1),利用同样的算法,估测的对应分子量分别为221 k及9 k,合计估测线粒体基因组为239 k。
|
Figure 2 Kmer frequency distribution of filtered RS41’s chloroplast data |
.png)
|
Figure 3 Kmer frequency distribution of filtered RS41’s mitochondrial data |
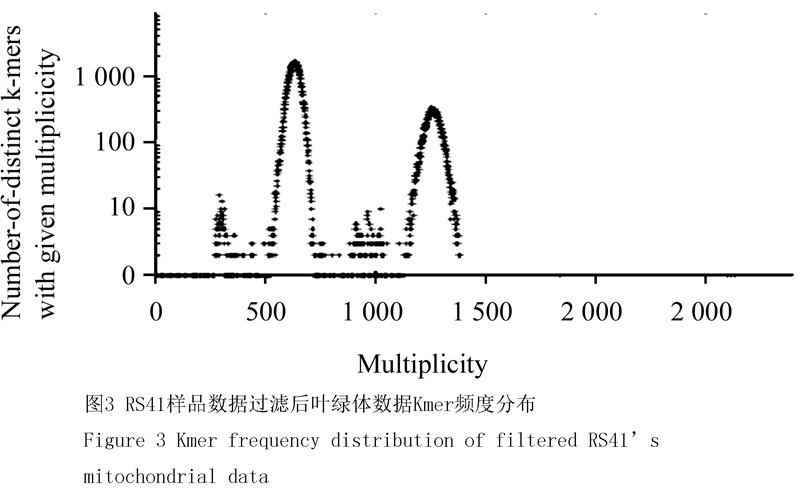
其余42、43、44、45四个样品进行的21mer,Kmer频度分析结果均与41号样品相似。预测的线粒体基因组240~250 k,预测的叶绿体基因组140~150 k,均符合预期。
Kmer分析表明,通过BWA比对及软件筛选搜集了来自不同细胞器的Reads。原始数据的分类过滤,有效提高了Reads的专一性。这为后续的组装打下了基础。
1.4叶绿体和线粒体基因组预组装
由于叶绿体的测序深度较高,我们利用全部Clean data组装叶绿体基因组。此时截止覆盖度高均高于来自线粒体与核数据的覆盖度,因此对其影响不大。组装后每个样品均得到6个Contigs,平均N50为55~58 k。对这些Contigs与NCBI公布的叶绿体基因组进行比对,结果表明所组装的Contigs与参照序列有很高的一致性,基本可断定为叶绿体基因组。
线粒体基因组组装,是叶绿体组装完成后,以叶绿体基因组为参照序列,从Clean data数据中严格滤除叶绿体的测序Reads,组装后,各样品包含32~40个Contigs,平均N50为40 k。对这些Contigs以NCBI公布的线粒体基因组为参考,进行比对,从中筛选出属于线粒体的Contigs,组装Contigs的统计结果见(表3)。
|
Table 3 The parameters for primary assembling and statistics on contigs |
.png)
1.5叶绿体和线粒体基因组的精细化
根据组装得到的Contigs,结合参照基因组设计引物进行PCR,对扩增产物进行Sanger测序,每个样品得到序列30~45条。利用Velvet软件结合第一次组装的Contigs,进行手动拼接。最后得到各自完整的全长基因组序列(表4)。
|
Table 4 Sum the genomes of chloroplast and mitochondria |
.png)
1.6基因组结构及基因注释
我们发现,叶绿体和线粒体基因组都呈现双环或三叶草型结构。都包含1个大单拷贝LSC,1个小单拷贝SSC及1对反向重复序列。基因注释结果表明它们包含了很多重要的自主功能蛋白、tRNA及rRNA的编码基因。
样品41为例,RS41萝卜叶绿体基因组全为153 445 bp,平均GC含量39.5%。它包括1个大单拷贝LSC,长度17 816 bp,1个小单拷贝SSC,长度83 197 bp和1对IR反向重复,长度26 216 bp。基因注释结果表明该叶绿体基因组包含87个编码蛋白的基因。均编码已知蛋白。在该基因组中共有外显子105个,平均长度为760 bp,总共占据基因组全长的51.7%。其中有72个蛋白不含内含子。而编码atpF、clpP、ndhA、ndhB、petB、petD、rpl16、rpl2、rpoC1、rps12、rps16及ycf3共12个基因包含了不止1个内含子。此外该叶绿体基因组还编码了20个tRNA的基因,其反密码子对应20种氨基酸。最后在rRNA方面,还包含了双份拷贝的16S、23S、4.5S及5S核糖体亚基的rRNA基因(图4)。
|
Figure4 Gene map for RS41’s chloroplast genome |
.png)
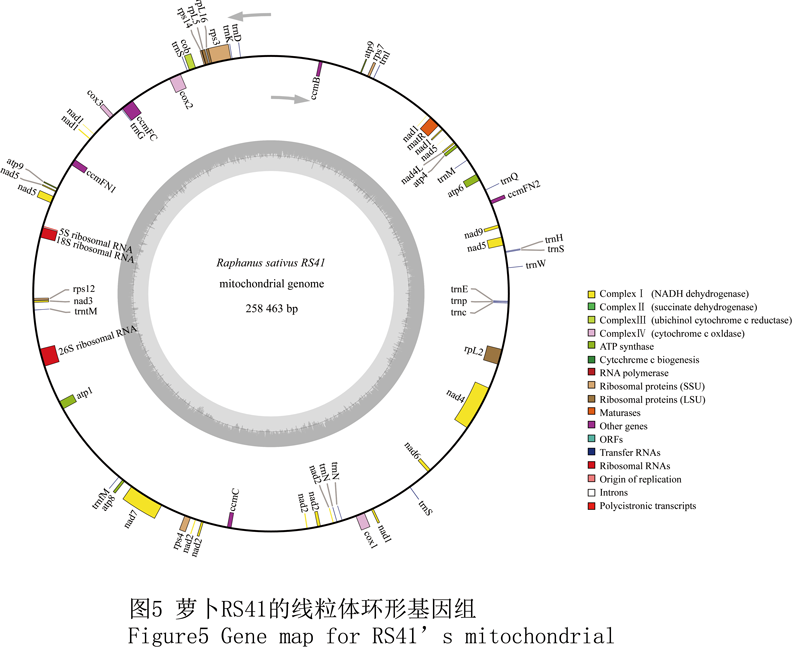
RS41萝卜线粒体基因组全为258 463 bp,平均GC含量为45.5%。它包括1个LSC单大拷贝,长度121 046 bp。1个小单拷贝SSC,长度117 955 bp。和1个IR反向重复,长度9 731 bp。基因注释结果表明该线粒体基因组包含82个编码蛋白的基因。其中34个为已知,而其它的48个ORF编码功能未知的蛋白。在该基因组中共有外显子105个,平均长度为482 bp,总共占据全长的19.5%。而编码rps3、cox2、ccmFC、nad1、nad5、nad2、rpL2、nad4和nad7的这9个基因包含了不止1个的内含子。此外该线粒体基因组还编码了17个tRNA基因,而这些基因对应14种氨基酸。它还编码了5S、18S及26S核糖体亚基的rRNA基因(图5)。其他样品基因组的基因注释结果均与此相似,不再赘述。
|
Figure5 Gene map for RS41’s mitochondrial |
2讨论
2.1DNA提取与文库构建
决定植物线粒体、叶绿体组装效果的关键因素在于细胞器DNA是否提取纯净,即完全的去除了不需要的核DNA成分。本文所采用的提取细胞器DNA的方法能够去除大部分的核DNA,但是很难分离线粒体和叶绿体。为此本文采用以参考基因组进行的比对、过滤方法来起到了分离线粒体和叶绿体DNA等价的效果。此时,Dry Lab的软件分析方法可以解决Wet Lab方法不足导致的实验缺陷。
另一方面,由于本研究是基于有参考基因组的组装。因此并未建立mate-pair文库、fosmid文库或进行三代Pacbio测序。如果是全新的De novo组装则必须有mate-pair文库、fosmid文库或三代测序的数据支持才能进行有效组装。因此应针对不同的组装方法采取不同的建库策略。
2.2线粒体与叶绿体的共有序列
在线粒体与叶绿体的组装中,我们发现两者存在6处大小不一的共有序列,这些共有序列存在较高的相似性。此处的Contigs难以确定归属,影响组装效果。特别是在线粒体组装,去除叶绿体Reads时应以本样品组装的叶绿体基因组为参考,进行严谨比对(mismatch=0),这样重复序列区段可以连接到线粒体基因组上,否则影响线粒体的组装。
2.3关于测序深度
从我们的组装情况看,对于像叶绿体和线粒体这样大小的基因组,能够达到800倍左右的测序深度,就比较好组装了,本研究的叶绿体组装就比较顺利,如果没有重复序列,组装的Contigs将会长而且数目少。而对于线粒体来说,测序深度仅110倍左右,组装难度大,组装出的Contigs多,存在较多Gap,如果没有比较相近的参照基因组,组装工作会非常困难。
2.4基因注释
组装完毕后我们对各基因组均进行了基因注释,基因注释的结果呈现出叶绿体、线粒体作为自主或半自主细胞器的特征(Ris and Plaut, 1962; Saccone et al., 2000)。而基因组之间的差异包括SNP及结构变异是否分布于CDS区域需要进一步的生物信息学分析来加以阐述。
3材料与方法
3.1实验材料
萝卜胞质雄性不育材料RS41、RS42、RS43,未知不育类型RS44及可育细胞质材料RS45由本课题组收集。试验材料种植于课题组试验田,田间管理按常规方法进行,待植株发育至10片叶时,取健康单株的顶部嫩叶20 g。
3.2线粒体、叶绿体提取
细胞器提取方法主要参考文献(曾秀存等,2005,田自华等,2004),每克材料加入5 mL匀浆缓冲液,在预冷的研钵中对材料进行研磨,保证整个过程温度不超过4℃,四层纱布过滤,收集滤液。滤液经2 000 g离心15 min后,取上清12 000 g离心10 min,取沉淀,上述离心过程重复两次,最终所得沉淀即为粗提线粒体和叶绿体。
3.3叶绿体及线粒体DNA提取
在纯化后的线粒体和叶绿体沉淀中加入2 mL预热CTAB缓冲液,65℃裂解1 h,12 000 g离心10 min,吸取上清液;加入等体积的苯酚-氯仿-异戊醇(25:24:1),混合摇匀,12 000 g,离心10 min,吸取上清;加入等体积的氯仿:异戊醇(24:1),混合摇匀,12 000 g,离心10 min,吸取上清;加入2/3体积的预冷异丙醇,冰浴30 min;12 000 g,离心1 min,用70%酒精洗涤沉淀数次;将DNA风干,加入30μL TE,-20℃保存备用。
3.4测序及数据质控与过滤
DNA经Illumina Standard Protocol,按PE90策略进行建库测序(https://support.illumina.com/ sequencing/protocols.html)。原始数据经去除测序重复,由Trimmomatic 3.0 (Bolger A., and F Giorgi., 2014)进行Clean处理后,用Fastqc (Andrews S., 2010)对Reads进行质控检测。以NCBI数据库(Boratyn., Grzegorz et al., 2013, Johnson et al., 2008)公布的萝卜核基因组,及5个萝卜线粒体为参考基因组,用五个样品的原始测序数据进行BWA (Li and Durbin 2009)比对(mismatch=0)。根据比对情况计算原始数据中细胞核、线粒体及叶绿体组分的比率。根据比对文件的Sam (Li et al., 2009)文件Mapping flag分值,将不能比对的数据过滤适合叶绿体组装用Reads,并转换为Fastqc文档。
待叶绿体组基因组装完毕之后,以各样本的叶绿体基因组为参考,用原始数据进行BWA比对(mismatch=0)。根据比对生成的Sam文件Mapping Flag分值,将不能比对的数据过滤出适合线粒体组装用Reads,并转换为Fastqc文档。
3.5Kmer分析及基因组组装
对过滤的数据分别进行Kmer分析,确定适合组装的mer值并预测测序深度及目标基因组的大小。
基因组大小的估测方法根据Marcai和Kingsford (2011)的方法,并加以改进。公式为:
.png)
.png)
用Velvet 软件(Zerbino and Birney., 2008)对过滤后的各样品数据进行组装,选择mer值为21,Insert Size为320,标准误sd为10,预期覆盖度、截止覆盖度参数则为表2计算得到的预计覆盖度。组装运行命令行为:
建立索引:Velveth ./Assem21 21 -shortPaired -Fastq in_fq1 in_fq2
组装Contigs:Velvetg ./Assem21 -exp_cov ${exp_cov} -cov_cutoff ${cutoff} -ins_length 320 -ins_length_sd 10
3.6提取Sanger测序及Gapping Filling
根据组装得到的Contigs的侧翼序列30 bp设计引物,以各样品在3.2.2提取的DNA样品为模板进行PCR扩增,并对扩增产物用Sanger法测序。
将第一步组装的Contigs序列及扩增产物经Sanger测序后得到的序列进行叠加,得到新的Scaffold,并结合IGV (Robinson et al., 2011)的可视化分析,进行手动拼接从而得到各自完整的全长基因组序列。最后用Mitofy (Alverson et al., 2010)软件对所有基因组进行基因注释。
作者贡献
段乃彬完成测序数据处理,基因组组装,基因注释及论文写作;王俊峰,白静及谢坤完成DNA提取,PCR扩增及Sanger测序以论文修改;王效睦是项目的构思者及负责人,指导实验设计,论文写作与修改定稿。全体作者都已阅读并同意最终的文本。
致谢
本研究由山东省自然科学基金(ZR2015YL054)及山东省农业良种产业化开发项目农业生物资源创新利用研究“萝卜地方品种雄性不育资源发掘与种质创新利用研究”共同资助。
Alverson, A. J., Wei, X., Rice, D. W., Stern, D. B., Barry, K and Palmer, J. D. 2010. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Molecular Biology and Evolution, msq029
http://dx.doi.org/10.1093/molbev/msq029
Andrews S., 2010, Fastqc: a quality control tool for high through put sequence data, Reference source,
http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Bolger A.M., Lohse M., and Usadel B., 2014, Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics.
http://dx.doi.org/10.1093/bioinformatics/btu170
PMid:24695404 PMCid:PMC4103590
Boratyn G.M., Camacho C., Cooper P.S., Coulouris G., Fong A., Ma N., and Zaretskaya I., 2013, BLAST: a more efficient report with usability improvements, Nucleic acids research, 41(W1): W29-W33
http://dx.doi.org/10.1093/nar/gkt282
PMid:23609542 PMCid:PMC3692093
Chen L., Liu L.W., Jin P., Gong Y.Q., Sun X.J., and Ma E.L., 2009, Cytological and molecular identification of cytoplasm in two male sterile lines in radish, Fenzi Zhiwu Yuzhong (Molecular Plant Breeding), 7(4): 757-762.(陈黎明, 柳李旺, 晋萍, 龚义勤, 孙新菊, 马二磊, 2009, 两个萝卜雄性不育材料胞质的细胞学与分子鉴定, 分子植物育种, 7(4): 757-762)
Cheng J.H., Li Y.C., Hu Q., Mei D.S., Li Y.D., Xu Y.S., and Wang W.M., 2008, Molecular identification and distinctness of N Sa male sterile cytoplasm in Brassica napus, Zuowu Xuebao (Acta Agronomica Sinica),34(11): 1946-1952 (程计华, 李云昌, 胡琼, 梅德圣, 李英德, 徐育松, 王巍敏. 2008, 油菜野芥 N Sa 细胞质雄性不育系的特异性分子鉴定, 作物学报, 34(11): 1946-1952)
Huang H., Liu L.W., Chen C.S., Gong Y.Q., Song X.Y., and Wei K.Y., 2004, Cytological investigation on microspore genesis of cytoplasmic male sterility in radish (Raphanus sativus L.), Zhiwu Yanjiu (Bulletin of Botanical Research), 24(3): 305-312 (黄浩, 柳李旺, 陈崇顺, 龚义勤, 宋贤勇, 韦开余, 2004, 萝卜 CMS 不育系与保持系小孢子发生的细胞学研究, 植物研究, 24(3): 305-308)
Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., and Madden T.L., 2008, NCBI BLAST: a better web interface, Nucleic acids research, 36 (supple2), W5-W9
http://dx.doi.org/10.1093/nar/gkn201
PMid:18440982 PMCid:PMC2447716
Li H., and Durbin R., 2009, fast and accurate short read alignment with Burrows–Wheeler transform, Bioinformatics, 25(14): 1754-1760
http://dx.doi.org/10.1093/bioinformatics/btp324
PMid:19451168 PMCid:PMC2705234
Li H., Handsaker B., W soker A., Fennell T., Ruan J., Homer N., and Durbin R., 2009, The sequence alignment/map format and SAM tools, Bioinformatics, 25(16): 2078-2079
http://dx.doi.org/10.1093/bioinformatics/btp352
PMid:19505943 PMCid:PMC2723002
Lin C., Sun P., Li X.F., Wu X., Chong F., and Gao J.W., 2007, Use of Cytoplasmic Male Sterile (CMS) Line to Produce Radish (Raphanus sativus L.) Hybrid F_1, Shandong Nongye Kexue (Shandong Agricultural Sciences), (4): 5-8 (林超, 孙萍, 李兴福, 吴雄, 程斐, 高建伟, 2007, 利用细胞质雄性不育系生产萝卜一代杂交种, 山东农业科学, (4), 5-8)
Marçais G., and Kingsford C., 2011, a fast lock-free approach for efficient parallel counting of occurrences of kmer, Bioinformatics, 27(6): 764-770
http://dx.doi.org/10.1093/bioinformatics/btr011
PMid:21217122 PMCid:PMC3051319
Ogura H., 1968, Studies on the new male-sterility in Japanese radish, with special reference to the utilization of this sterility towards the practical raising of hybrid seeds, Mem Fac Agric Kagoshima Univ, 6(2): 39-78
Ris H., and Plaut W., 1962, Ultrastructure of DNA containing areas in the chloroplast of Chlamydomonas, the Journal of Cell Biology, 13(3): 383-391
http://dx.doi.org/10.1083/jcb.13.3.383
Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., and Mesirov J.P., 2011,.Integrative genomics viewer, Nature Biotechnology. 29(1): 24-26.
http://dx.doi.org/10.1038/nbt.1754
PMid:21221095 PMCid:PMC3346182
Saccone C., Gissi C., Lanave C., Larizza A., Pesole G., and Reyes A., 2000, Evolution of the mitochondrial genetic system: an overview, Gene, 261(1): 153-159
Tian Z., Zhang Z., Zhang J., Shi S., Bai W., and Shao J., 2004, RAPD analysis of mitochondrial DNA of cytoplasmic male sterile line and it is maintainer line in sugarbeet, Fenzi Zhiwu Yuzhong (Molecular Plant Breeding), 2(6): 817-822 (田自华, 张子义, 张剑峰, 史树德, 白薇, 邵金旺, 2004, 甜菜细胞质雄性不育系与其保持系线粒体DNA的RAPD分析, 分子植物育种, 2(6): 817-822)
Zeng X.C., Sun W.C., Meng Y.X., Fan H.L., Wang B.C., Shao D.K., and Zhang J.W., 2005, Extraction and purification of mitochondria DNA in Crucifer, Xibei Zhiwu Xuebao (Acta Botanica Boreali-occidentalia Sinica), 25(6): 1137-1142 (曾秀存, 孙万仓, 孟亚雄, 范惠玲, 王保成, 邵登魁, 张金文, 2005.十字花科植物线粒体DNA的提取和纯化, 西北植物学报, 25(6): 1137-1142)
Zerbino D.R., and Birney E., 2008, Velvet: algorithms forDe novo short read assembly using De Bruijn graphs, Genome, research, 18(5): 821-829.
http://dx.doi.org/10.1101/gr.074492.107
PMid:18349386 PMCid:PMC2336801